製品技術資料作成支援
製品技術要求
「医療機器登録管理方法」及び関連規制は、医療機器の登録申請資料について新たに規定しました。医療機器安全有効基本要件リスト、統括資料、リスクマネジメント資料、研究資料、臨床評価資料等は追加要求されました。
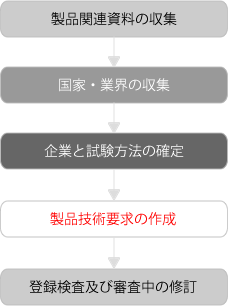
申請資料に最も重要な資料は製品標準から製品技術要求に変更したことです。製品技術要求は従来の記載方法と大きく変更され、製品標準より引用文献、製品部品や材料等の要求及び試験方法を削除され、完成品の要求や試験方法を記載することになります。一般的に、海外企業は自国や国際規格に基づき、製品自身の特性に合わせて製品の設計や生産したが、中国規格に製品に対して最低な要求を知らなかったことが多い。一旦、中国の最低規格要求に符合しない、製品はスムーズに承認を取得できない。しかし、海外企業は、中国の国家及び業界規格を収集及び理解するのは、言語、文字、考え方等の制限あります。従って、薬事申請専門者は製品技術要求が中国の国家業界規格に適合するかどうかを把握することが必要となります。
![]()
サービス内容
国家規格あるいは業界規格の調査
製品の特徴に基づき、中国に関連国家業界規格があるかどうかを調査します。国家規格あるいは業界規格がある場合、製品の状況に合わせて国家業界規格を採用でき、国家規格あるいは業界規格の関連内容を一部引用し、企業の製品技術要求を制定できます。
製品技術要求の編制
中国において、ほとんどの国家規格あるいは業界規格は直接採用できないため、自社の製品に合わせて、製品技術要求を作成しなければならない。
製品技術要求の確認
編制した製品技術要求を確認し、当該資料は国家規格あるいは業界規格または登録の要求に適合しているかを確定します。
![]()
![]()
ほかのサービス
リスクマネジメント資料の作成支援
臨床評価資料の作成支援(関連文献の調査、類似製品の調査、資料作成)
統括資料の作成支援
研究資料の作成支援(中国関連規制、規格、技術審査関連要求の提案)
型式試験関連資料の作成
その他
![]()
![]()
